Wanneer is een nieuwe techniek veilig genoeg?
1 reactie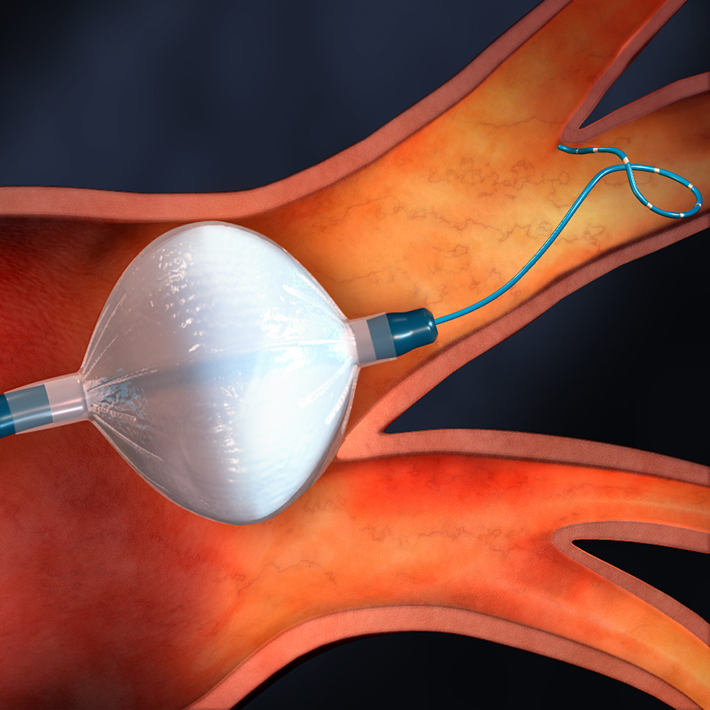
In het Erasmus MC gebruikten artsen de nieuwe cryoballonkatheter zonder hiervoor toestemming te vragen aan de patiënten, wat de nodige opschudding veroorzaakte. Hoe wordt er eigenlijk toegezien op de veiligheid van nieuwe hulpmiddelen?
De leidraad Nieuwe Interventies in de Klinische Praktijk uit 2014 van de Federatie Medisch Specialisten (FMS) en Zorginstituut Nederland bevat een stappenplan voor de introductie van een nieuw techniek. Eerste vraag: hoe nieuw is het eigenlijk? Experimentele zorg? Advies van de leidraad: niet invoeren en eerst maar eens praten met de wetenschappelijke vereniging. Nieuw in Nederland? Of nieuw in het eigen ziekenhuis? De leidraad adviseert: onderzoek of de vakgroep en het ziekenhuis toegerust zijn om ermee te werken. Maar voordat met het stappenplan de innovatieklasse kan worden bepaald, legt de leidraad in het voorwoord meteen de vinger op de zere plek: ‘In sommige gevallen is het gewoon lastig om te (h)erkennen dat er iets nieuws wordt geïntroduceerd.’
Lastig? ‘Ja, want hoe nieuw is nieuw?’, zegt cardioloog Lucas Boersma van het St. Antonius Ziekenhuis in Nieuwegein, die in de leidraadcommissie zat en NVVC-afgevaardigde is in de Raad voor wetenschap en innovatie van de Federatie Medisch Specialisten. ‘Voor de dokter is het altijd een vervolgstap op iets dat hij daarvoor al gebruikte. Maar je moet je als arts altijd afvragen: wanneer is iets zo nieuw – en daar is zo’n innovatiecommissie in het ziekenhuis voor om dat te beoordelen – dat je afstapt van de werkwijze tot nu toe en overgaat op een substantieel andere techniek waarvan nog niet zeker is of het helemaal veilig is.’
Innovatiecommissie
Zodra een producent een nieuw medisch hulpmiddel heeft ontwikkeld, vraagt hij er een CE-markering voor aan die de basisveiligheid voor de gebruiker moet garanderen en aangeeft dat het voldoet aan Europese regelgeving. In de ontwikkeling van het product tot dat moment is er niet noodzakelijkerwijs al een echte patiënt aan te pas gekomen bij wie het is uitgetest. Boersma: ‘Maar bij veel CE-trajecten in de medische wereld worden er ook pilotstudies onder enkele tientallen patiënten gedaan. Vaak doen firma’s vervolgens een limited market release in enkele ziekenhuizen om te zien of het goed gaat.’ In dat stadium komt de innovatiecommissie van het ziekenhuis eraan te pas, die zich bezighoudt met de beoordeling van alle nieuwe producten. Dat kunnen nieuwe hulpmiddelen bij bestaande ingrepen zijn, bijvoorbeeld een schaartje of een gaasje of iets groots als een apparaat. De leidraad vraagt vervolgens van artsen dat iedereen weet wat ze gaan doen, dat het ziekenhuis het goed vindt, dat ze die producten mogen inkopen en dat iedereen getraind is. Als artsen de leidraad van de FMS volgen en zich bekwaam achten, mogen ze de nieuwe techniek introduceren. Er is geen wet die voorschrijft dat medici volgen hoe het de patiënt vervolgens vergaat.
Aard van het beestje
Maar als het bedrijf of de arts een onderzoek wil koppelen aan de introductie, dan valt dit vaak onder de Wet medisch-wetenschappelijk onderzoek met mensen (WMO) en moet een erkende ethische commissie het beoordelen. Volgens Boersma is het de aard van het beestje dat artsen graag onderzoek doen naar een nieuw hulpmiddel. Boersma: ‘We staan voortdurend op de schouders van onze voorgangers, proberen heel snel te leren van de dingen die niet goed gaan en het handelen aan te passen. Zo gezien was het niet verkeerd dat ze in Rotterdam hebben gezegd: we gaan ook onderzoek doen of dit inderdaad veilig is en de gewenste resultaten heeft.’ Want al zijn de eerste resultaten veelbelovend, uiteindelijk moet in de praktijk blijken of die zich ook vertalen bij dagelijks gebruik. Boersma: ‘Die stap zul je altijd moeten maken en het is een lange weg van iets dat CE-approved is, naar de state-of-the-arttechniek. En wanneer is iets state of the art? Welke criteria hanteer je daarvoor? Wanneer is iets veilig genoeg? Waar moet je het mee vergelijken als iets totaal nieuw is? In Europa en in de meeste andere landen van de wereld, mogen spullen met een CE-markering commercieel in gebruik worden genomen. In Amerika heeft de FDA daar langdurige trajecten voor en ontwerpt samen met de firma’s en dokters protocollen. Uitgebreide klinische studies zijn verplicht, die gezamenlijk met de FDA worden opgezet, en waarbij aan zeer strenge criteria moet worden voldaan. Dat is wel een heel rigoureuze manier van testen, maar die kan ook een onwenselijke, forse vertraging opleveren. Ja, wat is beter?’
Mandarijnennetje
Volgens Maroeska Rovers, hoogleraar evidencebased surgery aan het Radboudumc moet er snel wat veranderen. ‘Het grote probleem is dat we nog steeds geen goed effectiviteitsonderzoek doen bij medical devices. Een innovatie hoeft nu alleen maar een CE-markering te hebben en het tv-programma Radar kreeg het keurmerk bijna voor een mandarijnennetje als bekkenbodemmatje. Veel mensen denken dat als iets een CE-markering heeft, het op veiligheid en effectiviteit is getest. Maar het hoeft dan nog niet in de patiënt te zijn getest. Er is dan gekeken naar veiligheid, maar niet per se naar de veiligheid ín de patiënt. En effectiviteit en werkzaamheid hoeven niet te zijn aangetoond. Dat is een prangend probleem, kijk maar naar de lekkende borstimplantaten, en de metaal-op-metaalheupen die metaalschilfers bleken te produceren.
Bij geneesmiddelen is dat totaal anders geregeld door de calamiteiten in de jaren zestig, toen duizenden kinderen werden geboren met afwijkingen doordat hun moeders tijdens de zwangerschap het nieuwe slaapmiddel Softenon hadden geslikt. Net als voor geneesmiddelen willen we ook voor hulpmiddelen beter toezicht. De Europese Unie heeft twee jaar geleden hierover een amendement aangenomen, maar moet nu nog met elke lidstaat apart afspraken maken. De neiging bestaat nu een beetje in Brussel om een soort kopie van het Europees geneesmiddelenagentschap EMA te maken als toezichthouder. Dan krijg je fase-I-, II- en III-onderzoek voor hulpmiddelen, maar daar ben ik niet voor, dan ben je zo vijftien jaar verder voordat je het in gebruik kunt nemen en sla je innovatie dood. Ik denk dat je al in een veel eerder stadium van ontwikkeling kunt kijken of een idee de zorg beter of goedkoper maakt. Je moet dan ook beter bekijken waaraan behoefte is. Artsen moeten daar natuurlijk bij betrokken zijn, en vooral ook de patiënt zelf.’
Minister Schippers over nieuwe medische hulpmiddelen
Minister Schippers beantwoordde vorige week Kamervragen van D66-Kamerlid Pia Dijkstra naar aanleiding van het NRC-artikel. Dijkstra vroeg of de minister het nodig vindt om nieuwe hulpmiddelen altijd zoals nieuwe geneesmiddelen te laten toetsen en beoordelen door een commissie. Dijkstra noemde de mandarijnennetjes waarvoor Radar bijna een CE-markering kreeg als voorbeeld. Schippers zei: ‘De systematiek die wij bij de geneesmiddelen hebben, kennen wij niet bij de hulpmiddelen, maar wij zijn het erover eens dat wij de wijze waarop we dit bij de hulpmiddelen hebben geregeld, onvoldoende vinden. U verwees al naar de netjes. Die zijn een enorme drijfveer geweest om tijdens het Europees voorzitterschap druk te zetten op de nieuwe regelgeving die wij vanuit Europa krijgen en die wij dus ook in Nederlandse regelgeving gaan omzetten. Die nieuwe regelgeving is veel strikter dan de huidige regelgeving. Daarin speelt klinisch onderzoek een veel belangrijkere rol. Het toezicht en de toelating zijn veel strikter geregeld. Er is meer aandacht voor bescherming van de proefpersonen die aan een klinisch onderzoek deelnemen. Kortom: die regelgeving hebben wij in ons voorzitterschap, na jaren vertraging, kunnen afronden. Eind dit jaar wordt die regelgeving vastgesteld en dan gaan we die ook in Nederland omzetten in wet- en regelgeving.’
NRC: Nieuw operatie-instrument getest op onwetende patiënten

















Ad J.J. Maas
klinisch fysicus, 's-Hertogenbosch
Het in het artikel geschetste probleem is reëel en actueel geworden nu binnen de ziekenhuizen steeds meer gekeken wordt naar de risico's die met de introductie van een nieuwe medische behandelmethode gepaard gaan. Een mogelijk criterium wordt verkreg...en door de vraag te stellen: "Wordt er een nieuw (bio)fysisch of (bio)chemisch principe toegepast?" Aanvullende criteria kunnen zijn, een risicoscore bij een gevalideerde risico screening (zoals in gebruik in het JBZ) en de extra kosten die aan de nieuwe behandelwijze vast zitten.
Dit thema lijkt mij een symposium waard. Door het uitwisselen van voorbeelden komen we dichter bij het juiste antwoord op de vraag: "Hebben we hier te maken met een innovatie of slechts een nieuwe verpakking?"