Hypoglykemieën en een micropenis
Plaats een reactieEen à terme neonaat heeft diepe hypoglykemieën en ontwikkelt ernstige hypotensie waarvoor frequente intravasale vulling en cardiotonica geïndiceerd zijn. Het kind heeft dysmorfe kenmerken en aangeboren afwijkingen waaronder micrognathie, postaxiale polydactylie, een atrioventriculair septumdefect (AVSD) en een micropenis met cryptorchisme. Er is geen hypospadie (figuur 1). Het chromosomenpatroon is normaal.
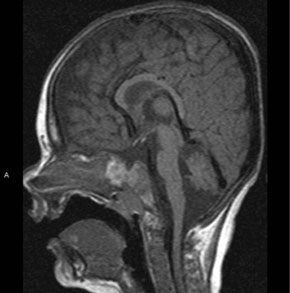
Er zijn geen aanwijzingen voor een sepsis, bloeding of cardiale oorzaak. Vanwege de micropenis en de lage bloedsuikerwaarde worden met spoed serum cortisol en ACTH bepaald. Er blijkt sprake van een centrale bijnierinsufficiëntie, waarop direct wordt gestart met hydrocortisonsuppletie. Hierop normaliseert de bloeddruk en doen zich geen hypoglykemieën meer voor. Omdat we denken aan panhypopituïtarisme worden ook de schildklierfuncties bepaald, die te laag zijn voor de leeftijd en passen bij een centrale hypothyreoïdie. MRI-onderzoek van de hypothalamus en hypofyse laat een lege sella turcica zien met een niet aangelegde adenohypofyse en een ectopische positie van de neurohypofyse net caudaal van het tuber cinereum. (figuur 2). Op de leeftijd van 2 maanden worden LH, FSH en testosteron bepaald, met uitslagen passend bij hypogonadotroop hypogonadisme. Ook is sprake van een groeihormoondeficiëntie. De diagnose panhypopituïtarisme, waarbij alle hormonen zijn uitgevallen, wordt gesteld.
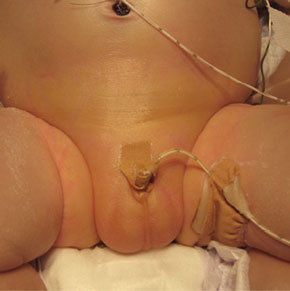
Panhypopituïtarisme in de neonatale periode kan zowel geïsoleerd als in combinatie met aangeboren afwijkingen voorkomen, vaak als onderdeel van een onderliggend syndroom. Het herkennen van het beeld is lastig en wordt bemoeilijkt doordat de symptomen weinig specifiek zijn en ook gerelateerd kunnen zijn aan prematuriteit en neonatale problematiek, zoals het optreden van hypoglykemieën, temperatuurinstabiliteit, cholestase en circulatoire insufficiëntie. Een micropenis met cryptorchisme kan duiden op hypogonadotroop hypogonadisme en vormt altijd aanleiding voor aanvullend endocrinologisch onderzoek.
Uiteindelijk werd bij het jongetje als oorzaak een mutatie in het GLI2-gen vastgesteld. Dit past bij het Culler-Jones syndroom, dat wordt gekenmerkt door hypofyseafwijkingen en polydactylie. Daarnaast zijn milde faciale dysmorfieën en een ontwikkelingsachterstand beschreven. Verschijnselen van holoprosencephalie komen zelden voor. De aandoening erft op autosomaal dominante wijze over en heeft een incomplete penetrantie en variabele expressie.
Omdat er nog niet veel patiënten met het Culler-Jones syndroom zijn beschreven, kunnen we niet veel zeggen over de prognose van dit kind. Het jongetje reageert goed op de behandeling van de hypofyse-uitval.
cc: redactie@medischcontact.nl
- Er zijn nog geen reacties















